Телефон
(831) 423-51-50
Адрес:
603107 г. Нижний Новгород, пл. Маршала Жукова, д.1
Пн-пт
07:00 - 20:00
Версия для слабовидящих
Вопросы и ответы
Женское здоровье
Заболевания и лечение
Снижение веса
Женское здоровье
Тянущие боли перед родами: причины и симптомы
Подробнее
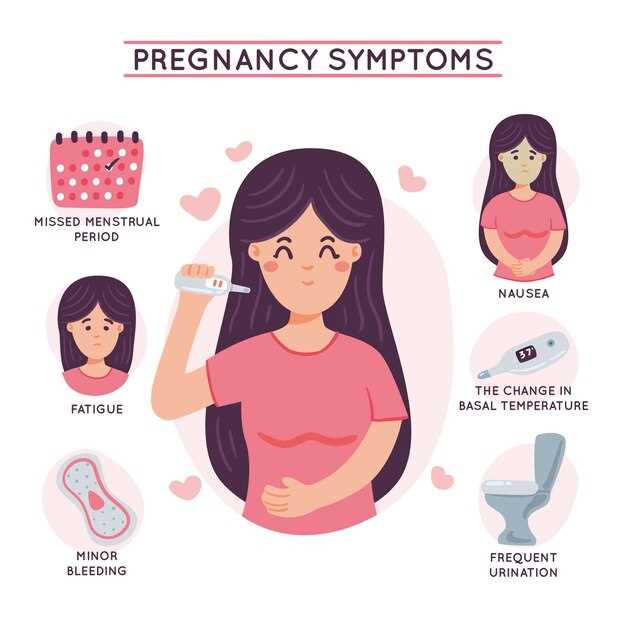
Тест на беременность вечером: эффективность и достоверность
Подробнее
Сон на животе при беременности: преимущества и рекомендации
Подробнее
Симптомы ПМС или беременности: как отличить их друг от друга?
Подробнее
Симптомы внематочной беременности и их отличия от других заболеваний
Подробнее
Почему болит живот и бока в начале беременности?
Подробнее
Без рубрики
Что делать, если у ребенка болит живот: советы от педиатра
Подробнее
Цистон или Роватинекс: сравнение эффективности и преимуществ
Подробнее
Сравнение средств экстренной контрацепции: эффективность и безопасность
Подробнее
Сколько раз в минуту дышит здоровый человек: норма и влияние факторов
Подробнее
Секреты вечернего ухода за кожей тела и лица
Подробнее
Признаки первой менструации: все, что важно знать
Подробнее
Вопросы и ответы
Эффективная гимнастика при спондилоартрозе: рекомендации и упражнения
Подробнее
Чистка лица и баня: советы по подготовке и безопасности процедуры
Подробнее
Черные точечки на сосках: причины и эффективные методы решения проблемы
Подробнее
Уход за тонкой кожей лица: советы по предотвращению возрастных изменений
Подробнее
Уход за губами после увеличения губ гиалуроновой кислотой: советы и рекомендации
Подробнее
Советы и рекомендации по улучшению состояния кожи при псориазе
Подробнее
Заболевания и лечение
Сравнение холензим и аллохол: какой препарат эффективнее при лечении печени?
Подробнее
Список аутоиммунных заболеваний: симптомы, диагностика и методы лечения
Подробнее
Причины боли в животе, вздутия и непереносимости определенных продуктов
Подробнее
Повышен уровень ТТГ: причины, симптомы, диагностика и лечение
Подробнее
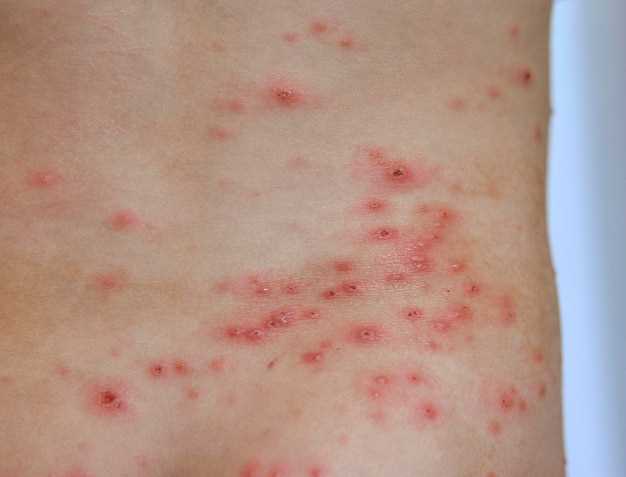
Красные точки на коже: причины, симптомы и методы лечения
Подробнее
Как узнать о начале овуляции: признаки и симптомы
Подробнее
Снижение веса
Диетолог дает советы по правильному питанию при непереносимости лактозы и молока
Подробнее
Диета при повышенном уровне тромбоцитов у женщин: советы по рациону и продукты, которые стоит включить
Подробнее
Диета при жировом гепатозе печени: меню на неделю с вкусными рецептами
Подробнее
Диета при гипертиреозе щитовидной железы: как правильно питаться для контроля заболевания
Подробнее
Диета при артрозе коленного сустава: эффективное меню на неделю
Подробнее
Диета на месяц: как быстро похудеть на 5 кг без ущерба для здоровья
Подробнее
Сейчас читают
Меню для пациентов с высоким уровнем креатинина: полезные продукты и рецепты
Подробнее
Красные точки на коже: причины, симптомы и методы лечения
Подробнее
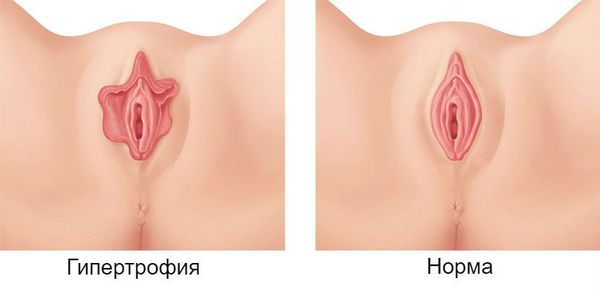
Гипертрофия малых половых губ: причины, симптомы и лечение
Подробнее
10 способов подтянуть кожу после резкого похудения
Подробнее
Микробная экзема: причины, симптомы и лечение
Подробнее
Вибрация во влагалище: причины, симптомы и методы лечения
Подробнее
1
2
3
Вперед »