Болезнь Фабри (болезнь Андерсона — Фабри) — это редкое генетическое заболевание, связанное с накоплением в организме жирового вещества, известного как глоботриаозилцерамид (Gb-3, GL-3, церамидтригексозид). Болезнь характеризуется возникновением новообразований кожи, боли в конечностях, плохого зрения, почечной недостаточности и заболевания сердца.

Кожные проявления болезни Фабри
Причиной болезни Фабри является мутация в гене GLA, локализованном в коротком плече Х-хромосомы (Хq22) . Мутация вызывает снижение фермента альфа-галактозидазы А, который отвечает за разрушение глоботриаозилцерамида.
Gb-3 относится к классу сфинголипидов — жиров, которые содержатся в оболочке (мембране) клеток и защищают клеточную поверхность. В норме они расщепляются и выводятся из клеток благодаря ферменту альфа-галактозидазы А, но при дефиците фермента Gb3 накапливается в клетках, начинает влиять на их нормальную работу и вызывает прогрессирующее повреждение организма, которое сопровождается разнообразными клиническими симптомами. Заболевание входит в группу сфинголипидозов, а сфинголипидозы относятся к группе лизосомальных болезней накопления, которые представляют собой редкие наследственные нарушения обмена веществ .
Распространённость болезни Фабри составляет от 1 на 40 000 до 1 на 120 000 новорождённых . Разница в данных объясняется трудностями постановки диагноза и разной частотой в популяциях.
Болезнь названа в честь дерматовенеролога Джон Фабри (J. Fabri), который в 1898 году впервые описал случай узелковой пурпуры и последовавшей за ней альбуминурией у тринадцатилетнего мальчика.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением — это опасно для вашего здоровья!
Симптомы болезни Фабри
Основные диагностические критерии:
- жгучие боли в дистальных отделах конечностей (ступнях и кистях);
- характерные кожные проявления;
- катаракта;
- кератопатия;
- накопление тригексозилцерамида в различных тканях;
- снижение активности фермента альфа-галактозидазы А в плазме крови, лейкоцитах, слёзной жидкости и культуре фибробластов .

Симптомы болезни Фабри
Первые признаки заболевания — постоянные нейропатические боли, резкие эпизоды акропарестезий (покалывания, и ощущения онемения в ступнях и кистях), непереносимость высоких и низких температур и чувство вялости — проявляются ещё в раннем школьном возрасте .
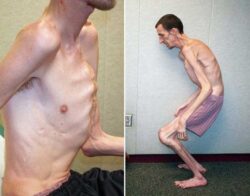
Больные мужчины характеризуются специфической внешностью: выступающие супраорбитальные дуги и лобные бугры, прогнатия (выдвижение вперёд верхней челюсти), пухлые губы и запавшая переносица .
Подробная характеристика клинических симптомов
Типичными для данного заболевания являются мучительные приступы акропарестезии. Больной при этом ощущает жжение, покалывание, дискомфорт в ступнях и кистях, которые возникают при небольшом болевом раздражении. Тяжесть таких приступов с годами значительно возрастает, приступы случаются чаще и становятся продолжительнее. Иногда приступы такой боли длятся по несколько дней, сопровождаются субфебрилитетом (повышением температуры тела в пределах от 37,1 до 38,0 °С), признаками воспаления по анализу крови . Для болезни Андерсона — Фабри характерно наличие болевых кризов — периодическое интенсивное нарастание болевых симптомов и их распространение на проксимальные отделы конечностей и туловище. Спровоцировать ухудшение состояния могут стрессовые состояния, перегрев, переохлаждение, изменения атмосферного давления, физическая нагрузка, усталость .
Ангиокератома является типичным поражением сосудов при болезни Фабри. Она представляет собой пятна вишневого цвета, слегка приподнятые над поверхностью кожи, безболезненные. Состоит из конгломерата нескольких расширенных сосудов, покрытых поверхностными слоями кожи. Локализуется на пальцах верхних и нижних конечностей, области вокруг губ, в районе гениталий, ануса, области вокруг пупка, на коленях. Не изменяет цвета при давлении. С годами возрастает их количество и размер. Ангиокератома обычно манифестирует в раннем возрасте и прогрессирует со временем .

Ангиокератома
Возможно развитие ангиэктазий (расширения просвета кровеносного сосуда) на слизистых оболочках полости рта и глаз .
К офтальмологическим симптомам относят увеличение диаметра и извилистый ход сосудов сетчатки и конъюнктивы, помутнение роговицы. При осмотре больных с помощью щелевой лампы иногда визуализируются светлые вихреподобные отложения субстрата в роговице, развивается воронкообразная кератопатия (патология роговицы, которая провоцирует снижение её прозрачности и нарушение зрения). В начальных стадиях заболевания острота зрения остается неизменной, но со временем развивается значительное помутнение роговицы, что может привести к слепоте. Поражение зрительного нерва происходит редко, но несёт за собой тяжёлые последствия.

Извилистый ход сосудов сетчатки и воронкообразная кератопатия
У пациентов с болезнью Андерсона — Фабри наблюдается нарушение потоотделения по типу гипогидроза (пониженного потоотделения), а иногда и ангидроза (отсутствия потоотделения). Это может приводить к непереносимости высоких температур, явлению перегрева организма.
Многие больные, особенно мужчины, жалуются на плохую переносимость физических нагрузок и утомляемость, что связано с быстрым перегревом и усилением интенсивности парестезий .
С возрастом в патологический процесс вовлекается сердечно-сосудистая система. Скопление сфинголипидов в мышечных клетках сердца и эндотелии (внутреннем слое) сосудов нарушает их функции и приводит к развитию фиброза (разрастания соединительной ткани). Проявляется это чувством одышки, болями за грудиной, аритмиями, тахикардией, гипертрофией левого желудочка, выявляемой по УЗИ, МРТ и ЭКГ. Вовлечение сердечно-сосудистой системы является наиболее неблагоприятным развитием болезни.
Поражение почек выявляется у всех гемизигот (мужчин, имеющих единственную мутантную Х-хромосому). Прогрессирует ХПН, которая на начальных стадиях имеет скрытое течение, нет повышения артериального давления, выявляется нормальный или слегка повышенный уровень креатинина в сыворотке крови, небольшая протеинурия (белок в мочи). Со временем почечная недостаточность достигает терминальной стадии, возникает тяжёлая уремия (тяжёлое самоотравление организма, обусловленное почечной недостаточностью), артериальная гипертензия.
Одними из самых явных проявлений заболевания являются неврологические нарушения. Больные предъявляют жалобы на приступы головокружения, головные боли, потерю сознания. Из-за накопления сфинголипидов в лизосомах нервных клеток и эндотелии сосудов возникают ишемические состояния головного мозга, в результате чего у больных случаются транзиторные ишемические атаки, ишемические и геморрагические инсульты. Зачастую ишемический инсульт в молодом возрасте является единственным проявлением заболевания, особенно у женщин-гетерозигот (имеющих одну нормальную и одну мутантную хромосому) и больных с атипичным типом заболевания . В результате ишемических изменений головного мозга больные становятся забывчивыми, рассеянными, неряшливыми, страдают когнитивные функции.

Ишемический и геморрагический инсульты
Больные могут жаловаться на шум, звон в ушах, резкое снижение остроты слуха, доходящее до глухоты. Наблюдается нейросенсорная тугоухость.
Поражение желудочно-кишечного тракта проявляется болью в области живота, диспепсией (затруднённым и болезненным пищеварением), диареей, метеоризмом, запорами. У большинства больных эпизоды диареи сменяются запорами, развивается геморрой. Иногда возникают желудочно-кишечные кровотечения .
У части пациентов наблюдается лёгкая железодефицитная анемия, не требующая коррекции . У мужчин отмечается отставание физического и полового развития.
Зачастую пациентам с болезнью Андерсона — Фабри ставят диагноз «ревматическая лихорадка», что связано с эпизодами субфебрилитета и повреждением костной системы. Костные нарушения проявляются вовлечением в патологический процесс дистальных межфаланговых суставов, снижением минеральной плотности позвонков и асептическими некрозами головок бедренной и таранной костей.
При тяжёлом течении заболевания, мучительных парестезиях нарушается психо-эмоциональная сфера человека, что приводит к депрессивным и тревожным состояниям. В некоторых случаях при интенсивных болевых проявлениях болезни пациенты имеют склонность к суициду.
Патогенез болезни Фабри
Болезнь Фабри наследуется по Х-сцепленному рецессивному типу наследования с полной пенетрантностью (когда мутантный ген проявляет свое действие у каждой особи) и различной экспрессивностью у мужчин (гемизигот) . Хотя некоторые авторы предполагают Х-сцепленный доминантный тип с неполной пенетрантностью (ген проявляется не у всех особей). Тип наследования в настоящее время является темой для дискуссии у специалистов .

Мутантный ген в Х-хромосоме
У мужчин-гемизигот заболевание протекает с более выраженными симптомами, как и у женщин-гомозигот (имеющих две мутантные Х-хромосомы). У женщин-гетерозигот (с одной мутантной Х-хромосомой) зачастую обнаруживается атипичная форма болезни, либо они оказываются здоровыми .
Как уже было отмечено, заболевание вызывается мутацией в гене GLA, локализованном в коротком плече Х-хромосомы (Хq22). Этот ген кодирует синтез фермента альфа-галактозидазы А, ответственного за расщепление сфинголипида глоботриаозилцерамида (Gb-3). Gb-3 в своей структуре несёт терминальный остаток альфа-галактозила, для отщепления которого требуется фермент альфа-галактозидаза А. При дефекте гена, кодирующего этот фермент, происходит нарушение процесса расщепления, что сопровождается отложением гликосфинголипидов с терминальным α-галактозил остатком в клетках.

Нормальный процесс расщепления Gb-3 и патология
Сфинголипиды — это липиды, производные алифатических аминоспиртов. Когда процесс их расщепления нарушается, происходит накопление церамидтригексозида в лизосомах клеток. Избыток субстрата в клетках значительно нарушает их жизнедеятельность, что приводит к изменению функций и клиническим проявлениям заболевания, нарушается нервная передача и связь между клетками.
Накапливаясь в сосудистой стенке, почках и миокарде сфинголипиды запускают процессы фиброгенеза (образования своеобразной рубцовой ткани), итогом которых является органная недостаточность . При накоплении субстрата в эндотелиальной ткани сосудов происходит их сужение, наблюдается нарушение микроциркуляции, гипоксия тканей. Это основной механизм развития ишемических нарушений.
Накопление сфинголипида в нервных клетках приводит к их структурным нарушениям: увеличению числа кальциевых каналов, образованию патологических ноцицептивных связей, повышению возбудимости путей болевой чувствительности и развитию типичной симптоматики .
Развитие гипогидроза, как и ангидроза, связано с депонированем сфинголипидов в клетках потовых желёз, нарушенной иннервацией и уменьшенным кровоснабжением кожи .
Накопление тригексозилцерамида в миокарде вызывает развитие прогрессирующей гипертрофической кардиомиопатии, а при наличии сужения коронарных сосудов это приводит к ишемическим кардиальным симптомам, острому коронарному синдрому и развитию сердечной недостаточности .

Гипертрофическая кардиомиопатия
Предполагается, что усиление боли при физических нагрузках связано со спазмом суженных сосудов, который приводит к ухудшению трофики нервных волокон .
Отложение субстрата в роговице приводит к её помутнению, а поражение сосудов может способствовать ухудшению зрения, вплоть до слепоты.
Классификация и стадии развития болезни Фабри
Выделяют две основные формы болезни Фабри :
- классическую;
- атипичную.
Классическая характеризуется ранней манифестацией заболевания (в первом десятилетии жизни) и наличием характерных симптомов и осложнений.
При атипичной форме происходит изолированное поражение головного мозга (ранние инсульты), сердца и почек. В этом случае заболевание манифестирует в более позднем возрасте и представляет большие трудности для диагностики .
Некоторые англоязычные авторы выделяют ещё женскую форму заболевания. Она имеет более лёгкие проявления, начало в среднем на 5-10 лет позднее классической «мужской» формы.
Осложнения болезни Фабри
Со стороны нервной системы:
- ишемические инсульты в молодом возрасте, нередко являющиеся причиной смерти.
Со стороны сердечно-сосудистой системы:
- ишемические инфаркты;
- кардиомиопатии;
- аритмии;
- сердечная недостаточность, приводящая к смерти.
Со стороны почек:
- терминальная (угрожающая жизни) стадия почечной недостаточности, требующая трансплантации почек.
Другие системы:
- нарушение зрения и слуха, вплоть до глухоты;
- переломы костей;
- перегрев организма.
Смерть чаще всего наступает от уремии или ишемических поражений мозга и сердца на четвёртом десятилетии жизни .
Диагностика болезни Фабри
Ранняя диагностика очень важна для правильного и своевременного лечения поражённых органов и предотвращения осложнений .
Важным методом диагностики болезни Фабри является оценка генеалогического анамнеза пациента. При этом могут обнаружиться родственники со сходными симптомами, которые не знают о своём заболевании. Ген, ответственный за болезнь Андерсона — Фабри, может передаваться через много поколений, поэтому ближние и дальние родственники пациента могут также иметь это заболевание. Для определения риска наследования болезни необходимо собрать информацию о здоровье всех известных членов семьи .

Как наследуется болезнь Фабри
Предварительный диагноз ставится по результатам опроса, сбора жалоб пациента и оценки генеалогического анамнеза. Для верификации диагноза используют методы обнаружения субстратов и энзимов, а также ДНК-диагностику.
- Измерение активности альфа-галактозидазы А . При болезни Фабри активность этого фермента у представителей мужского пола всегда ниже нормы, а у женщин этот показатель бывает в пределах нормы или слегка снижен. Материалом для проведения исследования могут служить лейкоциты, плазма крови, слёзная жидкость, культура фибробластов.
- Наиболее точным методом диагностики является секвенирование ДНК гена GLA . На сегодняшний день описано более пятисот мутаций этого гена, приводящих к болезни Фабри. Использование данной методики ограничено её стоимостью. ДНК-диагностику целесообразно применять у женщин, т. к. у них определение активности альфа-галактозидазы А не всегда даёт достоверный результат, и у родственников больного, т. к. они могут являться носителями мутантного гена или иметь атипичную форму заболевания. Проведение данного анализа возможно в Центре молекулярной генетики в Москве.
- Количественное определение глоботриаозилцерамида. Этот метод применяется для наблюдения за состоянием пациентов и оценки эффективности лечения, а также для определения формы заболевания (типичная, атипичная). Материалом для исследования могут быть плазма крови или сухие пятна крови.
- В некоторых случаях проводится биопсия почки с целью обнаружения клеток, содержащих лизосомы с характерным субстратом.

Биопсия почки
Болезнь Фабри бывает очень трудно отличить от более распространённых заболеваний, и пациенты в течение долгих лет могут оставаться без верного диагноза. Дифференциальная диагностика болезни Фабри проводится с наследственной геморрагической телеангиэктазией, ревматической лихорадкой, болезнью Фордайса, Шиндлера и другими наследственными болезнями накопления .
Лечение болезни Фабри
Лечение болезни Фабри состоит в замещении дефицитного фермента с помощью ферментозаместительной терапии. Она проводится с помощью внутривенного вливания препарата. Обычно ферментозаместительная терапия используется вместе с различными методами лечения конкретных симптомов .
В России сегодня используются два препарата для ферментозаместительной терапии: «Фабразим» компании Джензайм и «Реплагал» компании Шайер. Эти препараты очень дорогие. Лечение больных оплачивается из средств федерального бюджета, так как заболевание относится к орфанным (редким). Существует закон об ускоренной процедуре исследования лекарственных препаратов, предназначенных для лечения таких болезней .
Мужчинам начало ферментозаместительного лечения рекомендуется сразу после установления диагноза . Для купирования боли применяются препараты из групп атиконвульсантов, антидепрессантов, анальгетиков, нестероидных противовоспалительных препаратов и наркотических анальгетиков . Больным показано применение антигипертензивных препаратов .
При развитии терминальной почечной недостаточности проводят трансплантацию почки. Не рекомендуется, чтобы донорами были родственницы больного . В качестве коррекции тугоухости показано использование слуховых аппаратов.Физическая активность пациентов с болезнью Фабри должна быть ограничена из-за возможного усиления симптомов и перегрева организма .
Ситуации, при которых нецелесообразно назначать ферментозаместительную терапию:
- беременность и лактация;
- наличие другого опасного для жизни заболевания, при котором прогноз от применения заместительной терапии не станет лучше;
- наличие серьёзных осложнений (ишемический инсульт, реанимационные больные).
Прогноз. Профилактика
При надлежащем лечении и вовремя начатой заместительной ферментативной терапии прогноз для больных благоприятный. При отсутствии лечения смерть от сердечно-сосудистых, неврологических и почечных осложнений наступает на четвёртом десятилетии жизни .
Профилактика заболевания заключается в пренатальной или предимплантационной (при ЭКО) диагностике наличия мутантного гена методом ДНК-диагностики. Кроме этого, возможно прерывание беременности по медицинским показаниям . Иногда проводится скрининговое исследование сухих пятен крови с определением активности альфа-галактозидазы А, но оно ограничено финансовыми возможностями региона.
Необходимо обследовать всех пациентов с признаками болезни Фабри и потенциальных носителей гена для своевременного назначения заместительной ферментативной терапии, предотвращения клинических проявлений болезни и её осложнений. Для этого существует программа, которая регламентирует не только забор и отправку материала в Центр молекулярной генетики, но и обеспечение больных препаратами заместительной терапии.
Список литературы
- Козлова С. И., Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. — М.: Т-во научных изданий КМК; Авторская академия, 2007. — 448 с.
- Федеральные клинические рекомендации по диагностике и лечению болезни Фабри. — М., 2013.
- Matt Demczko Болезнь Фабри (болезнь Фабри; диффузная ангиокератома туловища) // Справочники MSD. Пользовательская версия. — 2018.
- Mehta A., Ricci R., Widmer U., et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey // European Journal of Clinical Investigation. — 2004; 34 (3): 236–242.ссылка
- Juan M. Politei, Didier Bouhassira et al. Pain in Fabry Disease: Practical Recommendations for Diagnosis and Treatment // CNS Neurosci Ther. — 2016; 22(7): 568–576.ссылка
- Omid Motabar, Ellen Sidransky, Ehud Goldin, and Wei Zheng. Fabry Disease – Current Treatment and New Drug Development // Curr Chem Genomics. —2010; 4: 50–56.ссылка
- Stephen Waldek, Sandro Feriozzi. Fabry nephropathy: a review — how can we optimize the management of Fabry nephropathy? // BMC Nephrol. — 2014; 15: 72. ссылка
- Frank Weidemann, Maria D Sanchez-Niño et al. Fibrosis: a key feature of Fabry disease with potential therapeutic implications // Orphanet J Rare Dis. — 2013; 8: 116. ссылка
- A Review of Fabry Disease / B. Chan, D. N. Adam // Skin Therapy Lett. — 2018; 23(2): 4-6.ссылка
- Маартен Арендс, Кристоф Ваннер, Дерралинн Хьюз Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study // J Am Soc Nephrol. — 2017; 28(5): 1631-1641.ссылка