
Синдром Прадера — Вилли (СПВ, Prader-Willi syndrome) — это сложное генетическое заболевание, поражающее многие системы органов . Для него характерна выраженная мышечная слабость, общая задержка развития и ожирение, которое появляется уже в детском возрасте . Этот синдром является самой частой генетической причиной ожирения .

Синдром Прадера — Вилли [31]
Синдром Прадера — Вилли был впервые описан швейцарскими педиатрами и учёными Андреа Прадером (A. Prader) и Генрихом Вилли (H. Willi) в 1956 году .
Распространённость
Синдром Прадера — Вилли встречается во всём мире. Хотя данные о его распространённости различаются, это, вероятно, связано с использованием разных методов выявления, так как нет убедительных доказательств повышенного риска в конкретных странах или генофондах.
В США синдром Прадера — Вилли выявляют у 1 из 10 000–30 000 человек. В других странах распространённость СПВ колеблется от 1:8000 в сельской местности Швеции до 1:16000 в Японии, 1:16000 в Австралии и 1:45000 в Великобритании . Мальчики и девочки болеют СПВ примерно с одинаковой частотой .
Причины синдрома Прадера — Вилли
Синдром Прадера — Вилли связан с отсутствием или недостаточной работой некоторых генов (или их частей) на 15-й отцовской хромосоме . Примерно в 70 % случаев заболевание вызвано хромосомной делецией 15q11–13 (потерей участка в 15-й отцовской хромосоме) .
Большинство случаев синдрома Прадера — Вилли имеют случайный характер, т. е. не передаются по наследству .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением — это опасно для вашего здоровья!
Симптомы синдрома Прадера — Вилли
При синдроме Прадера — Вилли в 10–40 % случаев выявляют ягодичное предлежание плода — ребёнок лежит на ягодицах по направлению к родовым путям. Дети с этим синдромом обычно рождаются в срок, но имеют низкую массу тела .
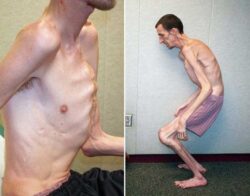
После рождения сразу наблюдается тяжёлая мышечная гипотония — пониженный тонус мышц. Она настолько выражена, что дети не могут совершать необходимые движения, даже сосать, что вызывает трудности с грудным вскармливанием. Таких детей приходится кормить с помощью зонда или пипетки. Кроме того, у маленьких пациентов резко задерживается формирование многих функций и навыков: они долгое время не держат или плохо держат голову, не сидят и не становятся на четвереньки. Физиологические рефлексы новорождённых при проведении неврологического осмотра могут не вызываться или быть сниженными. Также отмечается склонность к пониженной температуре (< 36,5 °C) .
Через несколько недель, месяцев или к концу первого — началу второго года жизни гипотония уменьшается и развивается булимия: ребёнок постоянно испытывает чувство голода и активно ищет еду, в результате чего возникает ожирение. При этом подкожно-жировой клетчатки больше всего на туловище, бёдрах и плечах.
Наблюдается акромикрия — стопы и кисти рук выглядят маленькими по сравнению с остальными частями тела.
Психическое развитие детей с синдромом Прадера — Вилли. У детей с СПВ имеется умственная отсталость, в особенности нарушено развитие речи: она затруднена, словарный запас чрезвычайно мал для конца первого и начала второго года жизни . Многие дети с этим заболеванием также отличаются особенностями поведения: обсессивно-компульсивным расстройством, перепадами настроения и упрямством. Последствия такого поведения проявляются в подростковом и взрослом возрасте и влияют на качество жизни, например наблюдается плохая успеваемость в школе и проблемы при общении со сверстниками .
Физическое развитие детей с синдромом Прадера — Вилли. Задержка роста является одним из главных критериев для постановки диагноза. Нарушение роста наблюдается в 60—90 % случаев СПВ . При рождении рост у таких детей соответствует норме или несколько ниже среднего (примерно 46–56 см). В дальнейшем его темпы снижаются, что особенно заметно в препубертатном и пубертатном периоде (примерно с 8–9 до 16–17 лет) . У многих пациентов с СПВ это связано с нехваткой гормона роста.
Также наблюдается нарушение работы гипоталамуса, который регулирует нейроэндокринную работу мозга и обмен веществ, что приводит к заболеваниям эндокринной системы, таким как гипогонадизм (отсутствие полового созревания в ожидаемое время: с 8–9 лет у девочек и с 10–11 лет у мальчиков), гипотиреоз и центральная надпочечниковая недостаточность. У мальчиков наблюдается гипоплазия (аномально малые размеры) полового члена и неопущение яичка (крипторхизм), а у девочек — гипоплазия больших и малых половых губ .
У пациентов с СПВ может развиваться апноэ сна и респираторные инфекционные заболевания (обструктивные бронхиты или пневмонии). Эти нарушения встречаются более чем в половине случаев и являются основной причиной гибели людей с СПВ .
Также может развиться остеопороз, при котором снижается минеральная плотность костей и возрастает риск переломов. Поэтому пациенты с синдромом Прадера — Вилли должны находиться под пристальным наблюдением эндокринолога на протяжении всей жизни .
Патогенез синдрома Прадера — Вилли
Долгое время тип наследования СПВ не был ясен. Многие учёные, которые впервые изучили и описали его, предположили, что наследование происходит по аутосомно-рецессивному типу. Но через некоторое время эта гипотеза была опровергнута наблюдаемыми случаями семейных патологий, которые указывали на аутосомно-доминантный тип наследования. Однако и эта гипотеза не подтвердилась, поскольку большинство случаев синдрома носили случайный характер. После открытия и внедрения в клиническую практику молекулярно-генетических методов удалось окончательно выявить изменения, происходящие в хромосомах людей с этим заболеванием, а именно повреждением участка 15-й хромосомы .
Развитие этого заболевания связано с двумя процессами, подробно изученных относительно недавно:
- геномным импринтингом, при котором активность генов некоторых участков хромосом различается в зависимости от их происхождения: материнского или отцовского ;
- унипарентальной дисомией — наследованием обеих хромосом только от одного родителя.
Открытие геномного импринтинга разрушило давнюю теорию о равном вкладе отца и матери в экспрессию генов.
Гены, ассоциированные с СПВ (область длинного плеча 15-й хромосомы, локусы 11–13) являются импринтированными и в норме экспрессируются только с отцовского аллеля. Гены такой же области 15-й хромосомы, полученной от матери, в норме не активны и не экспрессируются.
Экспрессия генов — это реализация заложенной в них информации, т. е. синтез РНК и белков. Другими словами, под экспрессией генов понимают их активность. СПВ возникает тогда, когда отсутствует экспрессия участка длинного плеча 15-й хромосомы (локусы q11–13), наследуемой от отца. Процессы импринтинга контролируются центром импринтинга, располагающимся в области 15-й хромосомы .
Таким образом, синдром Прадера — Вилли развивается из-за того, что определённые отцовские гены, которые должны экспрессироваться, отсутствуют по одной из следующих причин:
- 70 % случаев — микроделеция (потеря небольших участков) 15-й хромосомы, которая всегда имеет отцовское происхождение;
- 28 % случаев — материнская изодисомия, при которой ребёнок унаследовал две копии 15-й хромосомы от матери и ни одной от отца;
- 2 % — дефекты процесса импринтинга, какая-то ошибка или дефект в отцовских генах на 15-й хромосоме .
Несмотря на разные механизмы развития, клинические проявления не различаются .

Хромосомные нарушения при СПВ
Риски наследования синдрома Прадера — Вилли
Риск рождения ребёнка с такой патологией в семье, где уже есть один больной потомок, зависит от генетического механизма, который вызвал расстройство.
Вероятность рождения ребёнка с синдромом Прадера — Вилли:
- менее 1 % — при делеции гена или материнской изодисомии;
- 25 % — при появлении хромосомных транслокаций;
- до 50 % — при мутации региона, для которого характерно явление импринтинга .
Классификация и стадии развития синдрома Прадера — Вилли
В зависимости от особенностей питания выделяют четыре основные фазы СПВ:
I фаза. У ребёнка выражена мышечная гипотония, но нет избытка веса:
- I а — Проблемы при кормлении с нарушением глотания или без него.
- I б — Ребёнок ест, растёт и набирает вес нормально для своего возраста.
II фаза. Набор веса прогрессирует, обычно это происходит в период от 1,5 до 3 лет:
- II а — Вес увеличивается и превышает показатели, нормальные для возраста ребёнка, более чем в два раза. На этом этапе калорийность пищи ещё не меняется, т. е. ребёнок кушает столько, сколь ел до этого, но при этом набирает вес.
- II б — Ребёнок ест больше, что приводит к избытку веса или ожирению. Интерес к еде аномально повышен, но ещё может быть достигнуто насыщение.
III фаза. Начало весьма разнообразно, она может развиться как в ранние сроки — в 3 года, так и в поздние — в 15 лет. Характерен активный поиск пищи, отсутствие чувства насыщения, раннее появление чувства голода (в норме оно может возникать через 4–4,5 часа после еды, а у пациентов с СПВ — через 1–2 часа), из-за чего укорачиваются промежутки между приёмами пищи. При свободном доступе к продуктам пациенты могут съесть примерно в три раза больше, чем их сверстники без СПВ.
IV фаза. Высокий аппетит сохраняется, но он уже не столь выражен. Эта фаза развивается лишь у части взрослых пациентов.
Благодаря приёму гормона роста не все пациенты с СПВ проходят описанные этапы .
Осложнения синдрома Прадера — Вилли
Основные осложнения:
- нарушение углеводного обмена — у 25 % пациентов в возрасте 20 лет развивается сахарный диабет 2-го типа ;
- повышение артериального давления — гипертония встречается у 38 % взрослых с СПВ, но не характерна для детей .
Наиболее частые причины смерти пациентов с ожирением при СПВ:
- сердечно-сосудистая недостаточность;
- обструктивное или центральное апноэ;
- септицемия — одна из форм сепсиса, возникающая в результате попадания болезнетворных бактерий в кровяное русло; чаще всего развивается из-за кожных инфекций и пневмонии .
Серьёзные осложнения могут также возникнуть во время еды:
- Из-за быстрого потребления пищи высок риск удушья.
- При переедании, особенно у пациентов, которые временно остались без присмотра, есть риск некроза и разрыва желудка . Когда человек слишком много съел, стенки желудка сильно растягиваются, что приводит к нарушению кровоснабжения, из-за чего может развиться гипоксия, а затем некроз .
Диагностика синдрома Прадера — Вилли
Пренатальная диагностика проводится редко, но врач может заподозрить синдром Прадера — Вилли при уменьшении движений плода, неправильном предлежании и многоводии . Заболевание может быть выявлено при генетическом исследовании образцов, полученных при амниоцентезе (анализе амниотической жидкости из полости матки) .

Амниоцентез
Постанатальная диагностика. Синдром Прадера — Вилли можно предположить по симптомам и гормональным изменениям, но для подтверждения диагноза нужно обязательно пройти молекулярно-генетическое исследование, так как есть другие болезни с похожими проявлениями.
К таким заболеваниям относятся:
- синдром Барде — Бидля, для которого характерно ожирение, пигментный ретинит, полидактилия (наличие дополнительных пальцев на кистях или стопах), умственная отсталость, нарушение работы почек и гипогонадизм;
- синдром Альстрёма, при котором наблюдается врождённая дегенерация сетчатки, ведущая к слепоте, а также ожирение и сахарный диабет 2-го типа .
Показания для проведения молекулярно-генетического исследования :
| Возраст | Симптомы, достаточные для назначения дополнительного исследования |
|---|---|
| От рождения до 2 лет | Гипотония с нарушением сосания |
| 2–6 лет | Гипотония с нарушением сосания Задержка психомоторного развития Задержка роста и/или ускоренная прибавка веса |
| 6–12 лет | Гипотония с нарушением сосания Задержка психомоторного развития Избыточное питание (повышенный аппетит и одержимость пищей) с центральным ожирением, если приёмы пищи не контролируются |
| От 13 лет до взрослого возраста | Когнитивные нарушения — чаще всего лёгкое отставание умственного развития Избыточное питание Гипоталамический гипогонадизм и/или типичные поведенческие проблемы (приступы гнева и обсессивно-компульсивные расстройства) |
К показаниям для обследования также относятся специфические черты внешности: форма головы, при которой продольный размер значительно преобладает над поперечным, узкое височное расстояние, косоглазие, небольшой вздёрнутый нос с тонкой верхней губой и опущенными вниз углами рта.
Не всем детям с ожирением и проблемами с обучением нужно сразу проводить генетические исследования. Но ожирение — это уже повод обратиться к эндокринологу, который для начала осмотрит, соберёт анамнез (историю болезни) и жалобы, оценит гормональный статус. Если врач по собранным материалам заподозрит СПВ, то он отправит родителей и ребёнка на консультацию к генетику. Генетическое исследование также назначается подросткам и взрослым с поведенческими и психологическими проблемами, сочетающимися с ожирением и незавершённым половым созреванием .
Методы генетических исследований:
- Анализ метилирования ДНК – это единственный метод, который может подтвердить или опровергнуть диагноз СПВ, поэтому он проводится в первую очередь. Специфичность метода (вероятность, что тест правильно определит, что человек не болен) достигает 99 % . Если в результате теста обнаруживается только материнский импринт, СПВ подтверждается и рекомендуется провести микросателлитный анализ для выяснения механизма генетического наследования. Если наиболее частые генетические варианты СПВ (делеция, однородительская дисомия) не выявлены, желательно пройти дальнейшее исследование, чтобы исключить микроделецию центра импринтинга .
- Молекулярный микросаттелитный анализ гаплотипов — метод дополнительной диагностики, который назначают на втором этапе исследования при клинически предполагаемом диагнозе СПВ и исключённой микроделеции 15q11.2–q13. Проводится анализ наследования полиморфных ДНК-маркеров 15-й хромосомы для выявления заболевания, вызванного материнской однородительской дисомией .
- Метод флуоресцентной гибридизации in situ (Fluorescence in situ hybridization, FISH) — это вспомогательный метод диагностики. Из-за малого размера микроделеция не может быть выявлена с помощью стандартного анализа, но этот метод позволяет оценить наличие микроделеции на 15-й отцовской хромосоме. Обнаружение микроделеции — случайной мутации — позволяет прогнозировать низкий генетический риск повторения заболевания в семье .
- Мультиплексная лигазозависимая амплификация (MS-MLPA) — метод также является вспомогательным, благодаря ему можно выявить наличие и размер делеции на 15-й хромосоме .
Чтобы пройти генетическое исследование, достаточно сдать анализ крови.
Дополнительная диагностика:
- Анализ на ТТГ, свободный Т4 — позволяет оценить работу щитовидной железы, чтобы исключить гипотиреоз, особенно перед началом лечения гормоном роста.
- Печёночные пробы (АЛТ, АСТ) — у детей с СПВ и ожирением могут незначительно изменяться печёночные ферменты, что важно учитывать перед назначением гормона роста (чтобы понимать в динамике, будут ли какие-либо изменения при лечении).
- Сывороточный инсулиноподобный фактор роста-1 (ИФР-1) — для оценки дефицита гормона роста.
- Уровень глюкозы натощак и пероральный тест на толерантность к глюкозе — для обследования на сахарный диабет.
- Полисомнография (исследование сна) — для диагностики синдрома обструктивного апноэ сна.
- Двойная рентгеновская денситометрия (DXA-сканирование) — для определения минеральной плотности костей и состава тела (безжировой, жировой и тощей массы — той, что не относится к первым двум, преимущественно мышц и жидкостей) .
Лечение синдрома Прадера — Вилли
Заболевание нельзя вылечить, но можно облегчить состояние пациента. Чтобы стимулировать половое развитие, нужно принимать гормональные препараты . Чем раньше начнётся лечение, тем лучше для организма.
С момента постановки диагноза пациентам с синдромом Прадера — Вилли рекомендуется принимать препарат гормона роста. Это улучшает показатели роста и веса, повышает физическую силу и активность, что может предотвратить развитие ожирения . Но в любом случае ребёнку нужно соблюдать диету — за его аппетитом обязательно должны следить родители, а иногда и диетологи .
Возможные риски терапии гормоном роста:
- В начале лечения могут появиться расстройства дыхания, вызванные увеличением аденоидов и миндалин. Поэтому терапию начинают с меньшей дозы, затем в течение недель и месяцев её повышают, пока не будет достигнута стандартная заместительная доза. При приёме гормона роста рекомендуется проходить полисомнографию, а также оценивать состояние нёбных и носоглоточных миндалин как до, так и во время терапии гормоном роста (особенно в первые 3–6 месяцев).
- У детей с СПВ в 30–80 % случаев встречается сколиоз, связанный с мышечной слабостью и ожирением. Ускорение темпов роста, ожидаемое во время терапии, может неблагоприятно отразиться на состоянии опорно-двигательной системы. Такая терапия не влияет на развитие сколиоза и это нарушение не является противопоказанием для лечения, однако контролировать состояние опорно-двигательной системы на фоне лечения всё же желательно.
- Лечение гормоном роста способствует повышению уровня базального инсулина, развитию инсулинорезистентности и таким образом повышает риск возникновения диабета. В связи с этим рекомендовано систематически исследовать параметры углеводного обмена (контролировать уровень глюкозы, проводить стандартный пероральный глюкозотолерантный тест) как перед началом терапии, так и на фоне лечения гормоном роста .
Противопоказания для лечения гормоном роста:
- выраженное ожирение (вес в два и более раза превышает норму) в сочетании с апноэ сна, неалкогольной жировой болезнью печени или нарушениями обмена углеводов;
- апноэ тяжёлой степени .
Детям с отставанием психоэмоционального развития и задержкой речи потребуется психологическая помощь. В редких случаях (например, при появлении галлюцинаций, паранойи и депрессивного состояния) нужно обратиться к психиатру.
Советы по питанию при СПВ:
- есть максимально щадящую по консистенции пищу — по возможности жидкую, протёртую, пюреобразную и т. д.
- ежедневно есть суп на овощном или нежирном втором рыбном или мясном бульоне;
- ежедневно употреблять кисломолочные продукты без сладких наполнителей (ряженку, йогурт, кефир) — эти продукты необходимы для восстановления и поддержания здоровой желудочно-кишечной микрофлоры;
- ежедневно есть нежирное мясо, предпочтительнее диетические сорта мяса, птицы (фарш или мелкорубленное мясо);
- ограничить потребление сладких десертов: выпечку, конфеты и прочие кондитерские изделия;
- категорически исключить сахарозаменители;
- соблюдать чёткий режим дня и график приёма пищи;
- ужинать не более чем за 2–3 часа до сна;
- есть медленно, не отвлекаясь на телевизор, телефон и другие гаджеты;
- родителям не следует использовать еду как средство поощрения, шантажа или наказания;
- уделять внимание сервировке стола — одной из особенностей детей с СПВ является склонность к рутине и ритуалам: им нужно чётко знать меню, точное время приёма пищи или какого-либо события и важно, чтобы привычная последовательность дел строго выполнялась;
- хранить еду вне зоны досягаемости .
Прогноз. Профилактика
Прогноз зависит от степени развития осложнений и лучше при своевременной постановке диагноза и рациональной терапии . В настоящее время учёные ищут методы лечения, которые помогут улучшить прогноз для детей и взрослых с синдромом Прадера — Вилли .
Вовремя проведённое молекулярно-генетическое исследование помогает поставить диагноз на ранних сроках и своевременно привлечь врачей различных специальностей к лечению и реабилитации ребёнка . Поэтому при неправильном предлежании плода, его слабой подвижности и многоводии стоит провести генетическое исследование. Это не поможет предупредить развитие болезни, но позволит вовремя начать лечение.
Список литературы
- Prader-Willi syndrome // MedlinePlus. — 2022.
- Детская эндокринология. Атлас / под ред. И. И. Дедова, В. А. Петерковой. — М.: ГЭОТАР-Медиа, 2016. — 240 с.
- Гузева В. И., Бессонова Л. Б., Сеель К. А. Клинические трудности диагностики синдрома Прадера — Вилли // Педиатр. — 2013. — № 2. — С. 81–84.
- Goldstone A. P., Holland A. J., Hauffa B. P. et al. Recommendations for the Diagnosis and Management of Prader-Willi Syndrome // The Journal of Clinical Endocrinology & Metabolism. — 2008. — № 11. — Р. 4183–4197. ссылка
- Тозлиян Е. В. Синдром Прадера — Вилли в практике педиатра // Практика педиатра. — 2014. — № 2. — С. 32–39.
- Scheimann A. Prader-Willi Syndrome // Medscape. — 2021.
- Эллиот В., Эллиот Д. Биохимия и молекулярная биология: учебное пособие / под ред. А. И. Арчакова. — М.: Наука/Интерпериодика, 2002. — 430 с.
- Рыжкова А. Е. Этиология возникновения синдрома Прадера — Вилли // Матрица научного познания. — 2020. — № 11–2. — С. 29–34.
- Duis J. Prader-Willi syndrome: Clinical features and diagnosis // UpToDate. — 2023.
- Базарбекова Р. Б. Сидром Прадера — Вилли: клинический случай // Вестник КазНМУ — 2021. — № 3. — С. 191–194.
- Богова Е. А., Волеводз Н. Н. Синдром Прадера — Вилли: новые возможности в лечении детей // Проблемы эндокринологии. — 2013. — № 4. — С. 33–40.
- Driscoll D. J., Miller J. L., Cassidy S. B. et al. Prader-Willi Syndrome // GeneReviews® [Internet]. — 2023. ссылка
- Петеркова В. А., Васюкова О. В. Редкие формы ожирения // Лечащий врач. — 2008. — № 3. — С. 29–33.
- Казанцева Л. З., Новиков П. В., Семячкина А. Н. Синдром Прадера — Вилли у детей: новое в этиологии, патогенезе и лечении // Российский вестник перинатологии и педиатрии. — 1999. — № 4. — С. 40–44.
- Витебская А. В. Диагностика и лечение синдрома Прадера — Вилли. Обзор действующих международных консенсусов // Ожирение и метаболизм. — 2014. — № 3. — С. 9–17.
- Miller J. L., Lynn C. H., Driscoll D. C. et al. Nutritional phases in Prader-Willi syndrome // Am J Med Genet. — 2011. — № 5. — Р. 1040–1049. ссылка
- Butler J. V., Whittington J. E., Holland A. J. et al. Prevalence of, and risk factors for, physical ill-health in people with Prader-Willi syndrome: a population-based study // Dev Med Child Neurol. — 2002. — № 4. — Р. 248–255. ссылка
- Vogels A., Fryns J. P. Age at diagnosis, body mass index and physical morbidity in children and adults with the Prader-Willi syndrome // Genet Couns. — 2004. — № 4. — Р. 397–404. ссылка
- Tauber M., Diene G., Molinas C., Hébert M. Review of 64 cases of death in children with Prader-Willi syndrome (PWS) // Am J Med Genet A. — 2008. — № 7. — Р. 881–887. ссылка
- Whittington J. E., Butler J. V., Holland A. J. Pre-, peri- and postnatal complications in Prader-Willi syndrome in a UK sample // Early Human Development. — 2008. — № 5. — Р. 331–336.ссылка
- Glenn C. C., Deng G., Michaelis R. C., Tarleton J. et al. DNA methylation analysis with respect to prenatal diagnosis of the Angelman and Prader-Willi syndromes and imprinting // Prenat Diagn. — 2000. — № 4. — Р. 300–306. ссылка
- Butler M. G. Prader-Willi Syndrome: Clinical Genetics and Diagnostic Aspects with Treatment Approaches // Curr Pediatr Rev. — 2016. — № 2. — Р. 136–166.ссылка
- Powell-Hamilton N. N. Prader-Willi Syndrome // MSD Manual. — 2022.
- Gutierrez M. A. F., Mendez M. D. Prader-Willi Syndrome // StatPearls Publishing. — 2023.ссылка
- Пендина А. А. Болезни геномного импринтинга // Журнал акушерства и женских болезней. — 2007. — № 1. — С. 73–80.
- Богова Е. А. Клинические, генетические и гормонально метаболические особенности ожирения при синдроме Прадера — Вилли: дис. … к-та мед. наук: 14.01.02. — М., 2014. — 127 с.
- Юров И. Ю. Синдромы Прадера — Вилли и Ангельмана: возможности молекулярно-цитогенетической и цитогенетической диагностики // Журнал неврологии и психиатрии им. С. С. Корсакова. — 2014. — № 1. — С. 49–53.
- Хурс О. М., Политыко А. Д., Румянцева Н. В. Синдром Прадера — Вилли в Беларуси: генетическая структура и фенотипическая характеристика // Весці НАН Беларусі. Серыя медыцынскіх навук. — 2010. — № 1. — С. 5–10.
- Gourash L. M., Forster J. L. Prader-Willi Syndrome Food Security Basic Concepts. — Developmental and Behavioral Pediatrics Developmental Neuropsychiatry Pittsburgh Partnership, 2005. — 7 p.
- Stevenson D. A., Heinemann J., Angulo M. et al. Gastric Rupture and Necrosis in Prader-Willi Syndrome // J Pediatr Gastroenterol Nutr. —2007. — № 2. — Р. 272–274.ссылка
- Cortés F., Alliende M. A., Barrios A. et al. Paciente de 8 años con Síndrome de Prader Willi // Scientific electronic library online. — 2005.





