
Буллёзный эпидермолиз (Epidermolysis bullosa) — это болезнь, связанная с мутациями в разных генах. Степень тяжести и разнообразие клинических проявлений зависит от того, какой именно ген поражён.
Основной симптом — образование пузырей и эрозий на коже и слизистых оболочках, гиперчувствительность и ранимость кожи при незначительных механических травмах («механобуллёзная болезнь») .

Проявления буллёзного эпидермолиза [26]
Буллёзный эпидермолиз имеет две формы болезни: врождённую (ВБЭ) и приобретённую (ПБЭ) .
До 1886 года врождённый буллёзный эпидермолиз называли наследственной пузырчаткой. Впервые клиническую картину этой болезни описал британский доктор Дж. Хатчинсон в 1875 году .
При врождённом буллёзном эпидермолизе болезнь является результатом генной мутации, при которой дефектный ген передаётся от родителя к ребёнку . Установлено 18 генов, которые могут стать причиной болезни. Если один из них даёт сбой, кожа становится настолько непрочной, что даже лёгкое давление, трение или химическое и температурное воздействие могут стать причинами образования нового пузыря .
Приобретённый буллёзный эпидермолиз — хроническое аутоимунное заболевание, для которого характерно образование пузырей под слизистыми оболочками и наружным слоем кожи (эпидермисом). В этом случае нарушаются межклеточные связи в самом эпидермисе или между ним и дермой, в которой расположены сосуды, потовые железы и волосяные луковицы.

Образование пузырей между эпидермисом и дермой
Приобретённый буллёзный эпидермолиз проявляется хрупкостью кожи, пузырями, эрозиями и язвами на разных стадиях заживления кожи. Для него также характерно образование рубцов и белых угрей .
Распространённость буллёзного эпидермолиза
Долгие годы частота встречаемости врождённого буллёзного эпидермолиза была неизвестна, но после создания Национального регистра ВБЭ в США в 1986 году исследователи начали собирать и систематизировать данные о распространённости этой наследственной патологии. В Италии регистр пациентов с ВБЭ был сформирован в 1991 году. По состоянию на 2002 год там было зарегистрировано свыше 700 пациентов. Общая распространённость составила 10,1 случаев на 1 млн человек, а частота — 20,1 на 1 млн новорождённых. Регистры больных созданы также в Австрии, Австралии, Германии, Великобритании и других странах .
Статистический учёт больных буллёзным эпидермолизом в России отдельно не вёдется, поэтому точные данные о заболеваемости и распространённости неизвестны. Однако есть отдельные статистические данные, согласно которым средний показатель распространённости врождённого буллёзного эпидермолиза в России составляет 3,64 случая на 1 млн населения, а максимальный показатель распространённости приходится на Республику Дагестан — 19,73 случаев на 1 млн человек.
Высокий показатель распространённости (свыше 10 случаев на 1 млн населения) отмечают и в других регионах России, например в Томской области, Чеченской Республике, Республике Мордовии и Костромской области .
Среди зарегистрированных больных с врождённой формой болезни преобладают дети и подростки (как девочки, так и мальчики). Это связано с тяжёлым течением болезни и высокой смертностью пациентов до того, как им исполнится 18 лет. При этом совершеннолетние больные с лёгким течением заболевания редко обращаются за медицинской помощью .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением — это опасно для вашего здоровья!
Симптомы буллёзного эпидермолиза
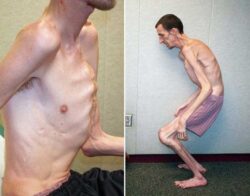
Основным клиническим признаком любой формы буллёзного эпидермолиза является появление пузырей при незначительной механической травме, например порезе, натирании одеждой, ссадине, расчёсе и т. д. Если повредить эти пузыри, образуется открытая рана, в которую может проникнуть инфекция.
К другим клиническим признакам относят огрубение и ороговение поверхности ладоней и подошв, а также нарушение пигментации кожи: на ней могут появиться как яркие пятна, так и участки с потерей пигмента (депигментация). Эти нарушения возникают на месте лопнувших пузырей.

Депигментация кожи
Скованность движений в суставах, сращение кожи между пальцами и отсутствие ногтей — частые проявления тяжело протекающих форм болезни.
Среди редко встречающихся признаков следует отметить выпадение волос, повышенное или недостаточное потоотделение, эрозии или пузыри в полости рта, кариес, сужение ротовой щели, затруднение глотания, рвоту, запор и диарею .
Даже незначительное увеличение температуры окружающей среды может ухудшить состояние кожи, провоцируя образование новых пузырей и/или эрозий.
Врождённая форма болезни проявляется с рождения или первых месяцев жизни и характеризуется непрерывным течением с периодическими обострениями.
Приобретённый буллёзный эпидермолиз у детей встречается чаще чем у взрослых. Клинические проявления этой формы напоминают течение буллёзного пемфигоида, когда к образованию пузырей присоединяется ещё и зуд с вовлечением слизистых оболочек. Пациенты с этой формой болезни обычно отмечают высокую эффективность монотерапии с использованием Дапсона .
Буллёзный эпидермолиз у взрослых часто сочетается с болезнями желудочно-кишечного тракта, например сужением просвета пищевода, анальными трещинами, запором, воспалительными болезнями толстого и тонкого кишечника, а также с системными аутоиммунными заболеваниями, сахарным диабетом и лимфомой .
Патогенез буллёзного эпидермолиза
Кожа человека состоит из эпидермиса, дермы и подкожно-жировой клетчатки. Эпидермис включается в себя пять различных слоёв эпителиальных клеток, или кератиноцитов, самый нижний из которых — базальный эпителий — прикрепляется к дерме с помощью множества разных белков. Эти белки не только определяют стабильность соединения клеток базального эпителия с дермой, но и их собственную прочность.
Врождённый буллёзный эпидермолиз является результатом мутации генов, которые кодируют структурные белки кожи. Снижение синтеза белков или полное его отсутствие приводит к разделению слоёв кожи и появлению пузырей. Уровень, на котором в коже формируется пузырь, и, соответственно, клиническая форма заболевания зависят от места, где находится дефектный белок .
Приобретённый буллёзный эпидермолиз появляется, когда организм сам разрушает коллаген VII типа. Этот тип коллагена отвечает за связь эпидермиса с дермой, в то время как сам коллаген — это белок, который обеспечивает прочность и эластичность соединительной ткани . Повреждение этой структуры нарушает связь эпидермиса с дермой и провоцирует спонтанное возникновение пузырей . Причина такого поведения организма до сих пор неизвестна.
Классификация и стадии развития буллёзного эпидермолиза
В соответствии с консенсусом Третьего международного согласительного совещания, принято выделять четыре основных типа врождённого буллёзного эпидермолиза:
- Простой буллёзный эпидермолиз — пузыри формируются на уровне или выше базального слоя эпидермиса. Они возникают в основном на открытых участках кожи, например стопах, кистях и локтях. Ногти, слизистые и зубы не включаются в процесс. Это лёгкая форма буллёзного эпидермолиза, при которой симптомы со временем исчезают и кожа заживает без рубцевания и омертвения (атрофии) тех участков, где прежде были пузыри. Более активно проявляется в детстве.
- Пограничный буллёзный эпидермолиз — пузыри образуются в верхней части базальной мембраны, которая соединяет два слоя кожи: эпидермис и дерму. В зависимости от степени тяжести пограничного буллёзного эпидермолиза клинические проявления разнятся от возникновения немногочисленных пузырей до обширного поражения кожи, угрожающего жизни больного. Обычно заживление пузырей происходит без образования рубцов, однако последующее заражение инфекцией может привести к рубцеванию и атрофии. При этой форме возможны аномалии зубов, выпадение волос и гиперпигментация кожи .
- Дистрофический буллёзный эпидермолиз — пузыри возникают на уровне нижней части базальной мембраны и в дерме, где находятся сосуды и нервы, поэтому пузыри бывают большими, болезненными и заполненными кровью. После них обычно остаются рубцы. Помимо рубцов для этой формы характерны белые угри, дистрофия и разрушение ногтей, ограничение подвижности пальцев, нарушение роста волос, деформация зубов, предрасположенность к плоскоклеточному раку кожи и задержка развития. Очаг поражения распространяется также на слизистые оболочки рта, желудочно-кишечный тракт и мочевые пути .
- Синдром Киндлера — пузыри формируются в эпидермисе на разных уровнях . Для этой формы характерны широкое распространение пузырей, светочувствительность, гиперпигментация кожи, внешне напоминающая сеть (пойкилодермия), дистрофия ногтей и атрофические рубцы. Зубы не подвергаются патологическим изменениям. Внекожные проявления включают в себя острый колит, эзофагит и др.

Пойкилодермия [23]
Среди клинических форм приобретённого буллёзного эпидермолиза выделяют два типа:
- Классический, или невоспалительный (механобуллёзный), эпидермолиз — проявляется хрупкостью кожи, пузырями и эрозиями, возникающими от незначительного механического воздействия либо после локальных травм, чаще в области костных выступов. Заживление приводит к образованию белых угрей и атрофических рубцов с нарушениями пигментации кожи, особенно у людей с тёмным типом кожи. Также возможно повреждение ногтей или их дистрофия.
- Воспалительный (пемфигоидоподобный) эпидермолиз — на фоне крапивницы, нормальной или покрасневшей кожи возникают зудящие, воспалительные, напряжённые пузыри, иногда заполненные кровью. При разрастании новообразований появляются эрозии, корки и изменение цвета кожи. Как правило, проходят без рубцевания .

Повреждение ногтей [24]
Описаны случаи развития одновременно классической и воспалительной форм ПБЭ .
Также учёные выделяют третий тип болезни, у которого нет официального названия. Он похож на рубцующийся пемфигоид, когда в патологический процесс вовлечены преимущественно слизистые оболочки с последующим их рубцеванием .
Ещё одна форма, при которой пузыри формируются под эпидермисом, напоминает течение линейного IgA-зависимого буллёзного дерматоза. Для него характерно поражение кожи туловища, рук, ног, лица и волосистой части головы, а также слизистых оболочек рта и глаз. У детей встречается редко .
Осложнения буллёзного эпидермолиза
Осложнения буллёзного эпидермолиза делятся на кожные и внекожные.
Кожные осложнения характерны для врождённого буллёзного эпидермолиза. К ним относятся:
- Большие пигментные родинки (невусы) от светло-бежевого до тёмно-коричневого и чёрного цвета, которые с возрастом светлеют.
- Повышенный риск меланомы у детей, даже на внешне нормальных участках кожи.
- Агрессивный, часто первично-множественный плоскоклеточный рак, устойчивый к химио- и лучевой терапии.
- Псевдосиндактилия, которой присуще не только сращение пальцев рук и ног, но и их атрофия, ограниченность или полная утрата движений большого пальца, кистей и стоп. Есть случаи рецидивов, которые возникают после операции, направленной на восстановление двигательной активности суставов.
Внекожные осложнения, характерные для обеих форм болезни:
- Поражение полости рта и желудочно-кишечного тракта, например тяжёлый кариес, ранняя потеря зубов, нарушение созревания твёрдых тканей зубов и частичное или полное отсутствие эмали, сужение просвета пищевода и заднего прохода, хронический запор, болезненная дефекация. В некоторых случаях пища или жидкость может попасть в нос, гортань и трахею, а глотание приносит боль.

Гипоплазия эмали [25]
- Поражение дыхательных путей, например отёк слизистых, пузыри, эрозии, рубцевание, охриплость голоса, сужение гортани, острая непроходимость дыхательных путей, которая сопровождается затруднением дыхания, одышкой и др.
- Поражение органов зрения, например эрозии и рубцевание роговицы, сращение одного или обоих век с глазным яблоком из-за появления пузырей на веках и слизистых, недостаточная выработка слёзной жидкости, воспаление краёв век и ухудшение зрения.
- Поражение мочеполовой системы, например болезненное мочеиспускание, сужение и непроходимость мочевых путей, пузырно-мочеточниковый рефлюкс, гидронефроз, почечная гипертония, гломерулонефрит, амилоидоз, почечная недостаточность и уросепсис.
- Ухудшение метаболизма и общего состояния (при тяжёлых формах буллёзного эпидермолиза). Из-за обнажения больших участков дермы организм расходует больше калорий, чем получает, поэтому появляется дефицит питательных веществ и белков. Ребёнок начинает отставать в росте, плохо себя чувствует из-за развивающейся анемии, у него медленно заживают раны и часто возникают инфекционные заболевания .
Диагностика буллёзного эпидермолиза
Диагноз «врождённый буллёзный эпидермолиз» традиционно устанавливают на основании симптомов и истории болезни, например по возникновению пузырных высыпаний или тенденции к их формированию с рождения, в младенческом или раннем детском возрасте. Чаще они появляются после незначительных физических и химических воздействий или спонтанно. Но клинических признаков и анамнеза недостаточно для постановки правильного диагноза — необходимо провести дополнительное молекулярно-генетическое исследование, которое выявляет мутацию генов, вызывающих патологию.
Диагностика врождённого буллёзного эпидермолиза
Симптомы врождённого буллёзного эпидермолиза могут отличаться даже в случае одной мутации или, наоборот, быть похожими при мутациях разных генов. Поэтому для установления субтипа болезни необходимо изучить биопсийный материал с помощью:
- трансмиссионной электронной микроскопии кожи — определяет уровень локализации пузыря и другие ультраструктурные изменения;
- прямой и непрямой иммунофлуоресцентной микроскопии — помимо местонахождения пузыря выявляет молекулярный дефект, лежащий в основе заболевания, т. е. наличие, отсутствие или недостаточный синтез искомого белка;
- генетической диагностики — обнаруживает мутантный ген .
Эти исследования дают возможность уточнить диагноз врождённого буллёзного эпидермолиза и определить его клиническую форму . Однако такие обследования можно провести не в любой клинике.
Диагностика приобретённого буллёзного эпидермолиза
С учётом неоднородности клинических проявлений диагностика приобретённого буллёзного эпидермолиза, основанная исключительно на внешних симптомах, затруднительна. В связи с этим применяют следующий алгоритм поиска:
- Гистологическая картина — изучение образцов тканей тела под микроскопом. В этом случае проводится гистологическое исследование небольшого кусочка кожи, взятого с помощью биопсии из очага поражения.
- Иммунофлюоресцентное исследование — с помощью флуоресцентных маркеров выявляет наличие антигенов. У пациентов с ПБЭ находят более широкую полосу отложения иммуноглобулина G между соединением дермы и эпидермиса, чем при других субэпидермальных аутоиммунных заболеваниях, сопровождающихся образованием пузырей, например буллёзном пемфигоиде .
- Электронная микроскопия околоочаговой зоны — определяет точное расположение волокон коллагена, которые организм пытается уничтожить, т. е. электронная микроскопия кожи проводится только при подозрении на приобретённый буллёзный эпидермолиз .
Лечение буллёзного эпидермолиза
Пациентов с подозрением на буллёзный эпидермолиз на всех этапах лечения консультируют дерматолог и генетик.
Лечение врождённого буллёзного эпидермолиза
Лечение врождённого буллёзного эпидермолиза в первую очередь основано на симптоматических методах. Симптоматическое лечение предотвращает образование пузырей или снижает их количество с помощью различных защитных средств.
Основной целью ухода за ранами является заживление кожи и слизистых оболочек. Большинство ран больных буллёзным эпидермолизом покрывют специальными атравматичными неприлипающими материалами (специальными пластырями), а после накладывают несколько слоёв бинтов. В некоторых случаях рекомендуют хирургическую обработку ран, например при их инфицировании.

Покрытие поражённых участков [16]
Для исправления деформаций кисти и стопы может потребоваться хирургическое вмешательство, например реконструктивно-пластическая операция или артродез (полное обездвиживание поражённых суставов).
В последние годы проводятся активные исследования возможностей генной терапии при лечении буллёзного эпидермолиза. Она основывается на попытках создать молекулярные конструкции, способные исправить работу мутантных генов, отвечающих за синтез структурных белков. Несмотря на достигнутые успехи, способ исправить наследственный дефект и излечить врождённый буллёзный эпидермолиз пока не найден .
Лечение приобретённого буллёзного эпидермолиза
Больных приобретённым буллёзным эпидермолизом лечат медикаментами, но чаще всего такая терапия неэффективна. Во многих случаях пациенты не реагируют на высокие дозы системных гормональных препаратов, например Азатиоприна, Метотрексата и Циклофосфамида.
Вместе с тем в ряде случаев показана эффективность монотерапии Дапсоном, а также его комбинации с Преднизолоном. Эти средства направлены на профилактику и лечение высыпаний и оказывают противовоспалительное, антибактериальное и противогрибковое действие.
Эффективен в лечении и Циклоспорин, однако токсичность препарата не позволяет широко его использовать .
Ещё одним методом является плазмаферез, при котором из плазмы удаляют токсичные вещества. Этот способ помогает добиться контроля над антителами к коллагену VII типа.
Отмечалась также эффективность использования внутривенного иммуноглобулина. В некоторых клинических исследованиях показаны положительные результаты применения:
- биологических препаратов против антигена фактора некроза опухоли-альфа (ФНОα);
- антител к CD-антигену против В-лимфоцитов, продуцирующих антитела .
Системная терапия при лечении буллёзного эпидермолиза
Также всем пациентам показана поддерживающая системная терапия, при которой следует:
- ухаживать за открытыми эрозивно-язвенными поражениями кожи, например обрабатывать антисептическими растворами и растворами с анилиновыми красителями;
- избегать травм, в том числе защищать участки кожи, наиболее подверженные травмам, использовать одежду без грубых швов и резинок, соблюдать охранительный режим и др.
Прогноз. Профилактика
Прогноз буллёзного эпидермолиза во многом зависит от подтипа болезни. Большинство пациентов с ВБЭ, особенно с простым и дистрофическим буллёзным эпидермолизом, могут с высокой вероятностью почти полностью избавиться от проявлений болезни и вести нормальную жизнь, хотя не исключена возможность возникновения серьёзных осложнений.
Напротив, для пациентов с пограничным буллёзным эпидермолизом прогноз неблагоприятный: большинство больных могут умереть в течение первых нескольких лет жизни, также они подвержены серьёзному риску осложнения в виде метастатического плоскоклеточного рака .
Пациенты с ПБЭ при правильном лечении и уходе имеют примерно такую же продолжительность жизни, как и здоровые люди. Прогноз одного ретроспективного анализа показал, что среднее время до ремиссии составляет 9 месяцев. Полная ремиссия через 1 год, 3 года и 6 лет наступила у 33, 33 и 45 % пациентов соответсвенно . Но даже при полной ремиссии для контроля болезни требуется длительная поддерживающая терапия.
Приобретённый буллёзный эпидермолиз редко приводит к смерти, но болезнь также может вызывать серьёзные осложнения. Кроме того, возможны проявления побочного эффекта от лекарств, используемых для лечения ПБЭ .
Профилактика буллёзного эпидермолиза
Чтобы оценить риск рождения ребёнка с буллёзным эпидермолизом и развития наследственного заболевания, проводится пренатальная диагностика — медико-генетическое консультирование семьи. Показаниями для консультирования являются:
- рождение ребёнка с врождённым буллёзным эпидермолизом;
- наличие болезни у одного из родителей;
- установленное или предполагаемое заболевание в семье.
Генетический анализ образцов крови и кожи больного позволяет уточнить тип и субтип врождённого буллёзного эпидермолиза, а также обнаружить мутацию соответствующего гена. Это очень важно для определения методов диагностики и тактики лечения болезни.
При беременности на сроке 10–12 недель проводят биопсию ворсин хориона с дальнейшим молекулярным исследованием полученного материала. Результаты получают в течение 3–4 дней после взятия материала. Это позволяет быстро проконсультировать семью и определить тактику ведения беременности.
Пациентам с врождённым буллёзным эпидермолизом для профилактики появления пузырей нужно беречь кожу и слизистые оболочки от травм, например носить свободную одежду, соблюдать диету, использовать нелипкие впитывающие повязки, ухаживать за полостью рта и т. д.
Для профилактики развития осложнений необходимо диспансерное наблюдение пациентов, в которое входит:
- периодическая сдача анализов для обнаружения и контроля анемии;
- полный осмотр больных для раннего выявления злокачественных опухолей кожи;
- своевременное лечение зубов .
В России есть только один фонд, который помогает людям с таким заболеванием — «БЭЛА. Дети-бабочки». Его участники помогают улучшить медицинскую помощь и всесторонне поддерживают людей с врождённой формой болезни.
Список литературы
- Fine J. D., Hintner H. Life with Epidermolysis Bullosa (EB): Etiology, Diagnosis, Multidisciplinary Care and Therapy. — New York: Springer, 2009. — 359 p.
- Вайс Х., Принц Ф. Реабилитационная терапия при буллёзном эпидермолизе / пер. с англ., под ред. Ю. Ю. Коталевской. — М.: Практика, 2015. — 190 с.
- Буллёзный эпидермолиз: руководство для врачей / под ред. Н. Н. Мурашкина, Л. С. Намазовой-Барановой. — М.: ПедиатрЪ, 2019. — 443 с.
- Кубанов А. А., Карамова А. Э., Чикин В. В. и др. Эпидемиология и состояние оказания медицинской помощи больным врождённым буллёзным эпидермолизом в Российской Федерации // Вестник РАМН. — 2018. — № 6. — С. 420–430.
- Mellerio J. E., Weiner M., Denyer J. E. et al. Medical management of epidermolysis bullosa: Proceedings of the 2nd international symposium on epidermolysis bullosa, Santiago, Chile // Int J Dermatol. — 2005. — № 8. — Р. 795–800. ссылка
- Laimer M., Lanschützer C. M., Nischler E., Klausegger A. et al. Hereditary blistering diseases. Symptoms, diagnosis and treatment of epidermolysis bullosa // Hautarzt. — 2009. — № 5. — Р. 378–388.ссылка
- Кубанов А. А., Альбанова В. И., Карамова А. Э., Чикин В. В. и др. Распространённость врождённого буллёзного эпидермолиза у населения Российской Федерации // Вестник дерматологии и венерологии. — 2015. — № 3. — С. 21–30.
- Кубанов А., Альбанова В., Чикин В., Епишев Р. Современные методы терапии врождённого буллёзного эпидермолиза // Вестник дерматологии и венерологии. — 2014. — № 6. — С. 47–56.
- Fine J. D., Bruckner-Tuderman L., Eady R. A. et al. Inherited epidermolysis bullosa: updated recommedations on diagnosis and classification // J Am Acad Dermatol. — 2014. — № 6. — Р. 1103–1126.ссылка
- Kindler T. Congenital poikiloderma with traumatic bulla formation and progressive cutaneous atrophy // Br J Dermatol. — 1954. — № 3. — Р. 104–111.ссылка
- Fine J. D., Eady R. A., Bauer E. A. et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB // J Am Acad Dermatol. — 2008. — № 6. — Р. 931–950.ссылка
- Megahed M. Histopathology of blistering diseases: with clinical, electron microscopic, immunological and molecular biological correlations. Textbook and Atlas 1st Edition. — Berlin: Springer, 2004. — Р. 223–235.
- Kablitz R. Ein Beitrag Zur Frage der Epidermolysis bullosa (hereditaria et acquisita): Dissertation. — 1904.
- Schachner L. A., Hansen R. C. Pediatric dermatology. — 4th Edition. — USA: Mosby Inc, 2011. — Р. 978–979.
- Мурашкин Н. Н., Опрятин Л. А., Материкин А. И. и др. Приобретённый буллёзный эпидермолиз у детей: серия клинических случаев // Вопросы современной педиатрии. — 2019. — № 1. — С. 56–64.
- Союз педиатров России. Врождённый буллёзный эпидермолиз: клинические рекомендации. — 2020. — 65 с.
- Mayuzumi M., Akiyama M., Nishie W., Ukae S. et al. Childhood epidermolysis bullosa acquisita with autoantibodies against the noncollagenous 1 and 2 domains of type VII collagen: case report and review of the literature // Br J Dermatol. — 2006. — № 5. — P. 1365–2133.ссылка
- Fine J. D. Inherited epidermolysis bullosa // Orphanet J Rare Dis. — 2010. — № 12. — Р. 1750–1172.ссылка
- Kim J. H., Kim Y. H., Kim S. C. Epidermolysis bullosa acquisita: a retrospective clinical analysis of 30 cases // Acta Derm Venereol. — 2011. — № 3. — Р. 307–312. ссылка
- Reinhart J. Epidermolysis Bullosa Acquisita // Medscape. — 2019.
- Intong L. R. A., Dеdее F. M. Inherited epidermolysis bullosa: new diagnostic criteria and classification // Clin Dermatol. — 2012. — № 1. — Р. 70–77.ссылка
- The underlying causes of prominent skin conditions // Victorian Dermal Group. — 2020.
- Femia A. N. How is poikiloderma in dermatomyositis characterized? // Medscape. — 2018.
- Гаджимурадов М. Н., Гаджимурадова К. М., Алиева М. Г., Мамашева Г. Д. Врождённый буллёзный эпидермолиз. Клинические особенности и собственные наблюдения // Клиническая дерматология и венерология. — 2020. — № 5. — С. 647–654.
- Penarrocha M., Martinez C. S., Silvestre F. J., Bagan J. V. Hereditary epidermolysis bullosa. Dental management of three cases // Medicina oral. — № 1. — Р. 48–56.
- Fine J.-D. Epidermolysis Bullosa Simplex (localized EBS; Weber-Cockayne; Dowling-Meara (EBS herpetiformis) // Dermatology Advisor. — 2013.



: причины, симптомы и лечение")

