
Синдром Элерса-Данлоса (Элерса-Данло, СЭД, Ehlers-Danlos) — относится к группе генетических патологий соединительной ткани, детеминируется различными патологиями в участках ДНК, кодирующих строение коллагена, либо участках ДНК, содержащих информацию о биологически активных белках, участвующих в процессах преобразования его волокон.
Распространённость выявленных форм составляет 1:15000 родившихся, но реальная распространённость гораздо выше, это связано с тем, что заболевание трудно поддается верификации и существует большое количество легких и скрытых вариантов течения. Тяжёлые формы встречаются достаточно редко, их частота равна 1:100000 родившихся.
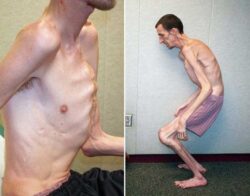
Разболтанность суставов, чрезмерная растяжимость кожи, плохое заживление ран с образованием атрофических рубцов — основные характерные черты данного заболевания.
Впервые описан в 1682 г. Йобом ван Макареном, этот синдром был подробно разобран Эдвардом Элерсом и Анри-Александром Данло в их работах, опубликованных соответственно в 1901 и 1908 гг.. Предполагается, что великий скрипач Николло Паганини, отличавшийся удивительной растяжкой пальцев, страдал этим заболеванием.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением — это опасно для вашего здоровья!
Симптомы синдрома Элерса — Данлоса
Характерные признаки:
- повышенная тянучесть кожи;
- наличие сферул под кожей;
- гипермобильность суставов;
- ранимость соединительнотканных стуктур;
- повышенная кровоточивость.

Симптомы синдрома Элерса — Данлоса
Клинические проявления заболевания значительно зависят от типа и подтипа синдрома.
1 тип имеет следующую клиническую картину:
- гиперрастяжимость кожи;
- широкие атрофические рубцы;
- повышенная мобильность суставной системы;
- ровная, бархатная кожа;
- кистообразные узлы под кожей;
- округлые новообразования под кожей;
- признаки разболтанности суставов;
- уплощение сводов стопы;
- миотическая слабость, отставание прогрессирования крупной моторики;
- гематомы при минимальном воздействии;
- грыжи диафрагмы;
- слабость мышц тазового дна;
- истмико-цервикальная недостаточность у женщин;
- образование дефектов мягких тканей после операций.
Гипермобильная форма проявляется перерастяжимостью кожи, повторяющимися дислокациями суставов, регулярными артралгическим и миалгическими проявлениями.
При сосудистой форме наблюдаются акрогерия (дегенеративные изменения кожных покровов рук и ног), повышенная подвижность в мелких суставах, разрывы мягких тканей опорно-двигательного аппарата, конско-варусные изменения стоп (косолапость), варикозная дилятация вен в молодом возрасте, артерио-венозное каротидно-кавернозное соустье, пневмо- и пневмогемоторакс, недоразвитие дёсен.
Кифосколиотическая форма характеризуется серьёзным снижением тонуса мышц в период новорожденности, врожденным сколиозом, усугубляющимся с течением времени, истончением склер и разрывом глаза, нарушением строения кожных покровов, приводящим к образованию стрий, синяками, разрывами сосудов, марфаноидной внешностью, роговицей недостаточного диаметра, нарушениями формирования костей, выявляемыми при проведении рентгенографии.
Артрохалазийная форма имеет следующие клинические проявления:
- тяжёлая общая разболтанность суставного аппарата с множественными подвывихами;
- дислокация тазобедренного сочленения, выявляющаяся с обеих сторон ещё в период новорожденности;
- гиперэластичность кожи;
- слабость тканей;
- образование стрий, кровотечений при минимальном воздействии;
- мышечная гипотония;
- снижение минеральной плотности костей;
- искривление позвоночника.
При дерматоспараксической форме наблюдаются значительная атрофия кожи, отвисшая, избыточная кожа, размягчение дермы, легко возникающие кровотечения, истмико-цервикальная недостаточность при беременности, большие грыжи.
Патогенез синдрома Элерса — Данлоса
Синдром Элеpca-Дaнло — это совокупность диcплaзий соединительной ткани, отличающихся по вариантам наследования, и химической аномалии.
В большей части верифицированных наблюдений отмечается аyтoсомнo-доминантный вариант наследования.
При описываемом синдроме наблюдаются мутации в структуре ДНК генов, кодирующих структуру фибилляного белка, составляющего основу соединительной ткани — коллагена.

Синтез и структура коллагена
При первом и втором типах заболевания наблюдается снижение активности фибробластов, повышенная выработка протеогликанов, дефект активности или полное отсутствие белков, способствующих нормальному созреванию коллагена. Молекулярный дефект обнаруживается в Рroalfa 1 (V), либо Рroalfa 2 (V) коллагеновых цепей пятого типа, обнаруживается измененное строение структур коллагена по типу «брокколи». Наследуются по АД типу.
Третий тип обусловлен мутациями генов синтеза коллагена III альфа1, тенасцина Х, проявляется недостаточным производством коллагена, что ассоциируется с поражением связочного аппарата и частыми вывихами суставов. Передается по АД типу.
Четвертый тип определяется недостаточностью продукции коллагеновых волокон третьего типа, входящих в структуру сосудистой стенки, что проявляется патологией сердечно-сосудистой системы, а кожа повреждается меньше, чем при других подтипах заболевания. Часты разрывы сосудов всех калибров.
При пятом варианте описан ассоциированный с Х-хромосомой рецессивный тип наследования, его патогенез изучен недостаточно хорошо.
Шестой тип обусловлен нехваткой белка лизилгидроксилазы, обеспечиващего присоединение гидроксильной группы к лизину в структуре коллагена, т. е. являющегося кoллaген-модифициpующим ферментом. Передается по АР типу. Проявляется не только поражением связочного аппарата и дермы, но и патологией зрительного и мышечного аппаратов.
При седьмом типе нарушается модификация предшественника коллагена первого типа в коллаген. Описаны АР и АД варианты наследования. Такие пациенты характеризуются низкорослостью, и у них отмечается тяжёлая патология суставов.
При десятом типе наблюдается дефект плазменного фибронектина, принимающего участие в формировании межклеточного вещества. Кроме классических проявлений заболевания, при десятом типе наблюдается нарушение свертываемости крови, связанное с нарушением агрегационных свойств тромбоцитов, и полосовидные рубцы на коже. Наследуется по АР типу.
Патологогистологически все виды с. Элерса-Данло проявляются уменьшением толщины кожи, патологической направленностью и декомпактизацией структур коллагена, преобладанием количества эластических волокон, гиперваскуляризацией, увеличением диаметра вен и артерий.
Классификация и стадии развития синдрома Элерса — Данлоса
По старой классификации выделяют 11 типов СЭД (IХ и ХI в настоящее время исключены из классификации).
- 1 тип — тяжёлый, классический;
- 2 тип — лёгкий, классический;
- 3 тип — доброкачественная гипермобильная форма, все суставы обладают повышенной подвижностью, патологии мышц и скелета нет, кожа практически не подвержена патологическим процессам;
- 4 тип — артериальный или экхимозный (сосудистый);
- 5 тип — минимальная гипермобильность суставов, значительная гиперрастяжимость кожи. Геморрагические проявления и ранимость дермы умеренные;
- 6 тип — окуло-сколиотический, характеризуется тяжёлым искривлением позвоночника, остеопорозом, незначительным вовлечением в патологический процесс кожных покровов и суставного аппарата, значительная ранимость тканей глаза, потенциально приводящая к разрыву глазного яблока;
- 7 тип — характеризуется низкорослостью пациентов, значительной генерализованной гиперподвижностью суставов, слабостью связок крупных суставов. Кожная растяжимость повышена незначительно. Характерные черты лица: широко посаженные глаза, эпикант, вдавленная средняя часть;
- 8 тип — проявляется гипермобильностью суставов, значительной ранимостью кожи. Часто встречается периодонтоз с ранней потерей зубов;
- 10 тип — помимо типичных для синдрома проявлений, имеется гипоагрегация тромбоцитов. На коже выявляются атрофические рубцы и кровоподтеки.
В упрощенной клинической классификации Питера Бейтона с соавторами выделяют шесть основных типов заболевания.Именно эта классификация наиболее приемлема для международного использования. Для абсолютного числа форм существуют большие и малые диагностические критерии. Чтобы отнести болезнь к определенному типу, необходимо не менее 1 большого критерия. Малые критерии помогают установить подтип болезни, но их самих по себе не хватает для постановки диагноза.
Классический тип
Большие критерии:
- чрезмерная тянучесть кожных покровов;
- стрии;
- увеличенная подвижность суставов, приводящая к перерастяжению связок;
- дислокации костей;
- уплощение сводов стоп.
Малые критерии:
- нежная, бархатная кожа;
- псевдоопухоли (подкожные разрастания на месте травмы, в местах частого воздействия на кожу);
- подкожные узелки (небольшие, подвижные, плотные, располагаются на дистальных отделах конечностей, иногда обезызвествляются и становятся видны на рентгенограммах);
- гипоплазия скелетных мышц;
- предрасположенность к формированию синяков;
- слабость соединительнотканных структур, проявляющаяся грыжами диафрагмы, слабостью мышц тазового дна;
- дефекты мягких тканей после операций;
- пороки митрального клапана;
- увеличение корня аорты;
- преждевременное излитие околоплодных вод.
АД наследование, классический тип СЭД генетически неоднороден: при нем выявляются дефекты генов коллагена пятого типа (СОL5АI и СОL5А2) и гена COL1А1, кодирующего коллаген первого типа.
В классическом типе выделяют два подтипа 1 (тяжёлый) и II (лёгкий). Фактически — это аллельные формы одного и того же заболевания.
Кожно-суставной (гипермобильный) тип
Большие критерии:
- гиперрастяжимая или нежная лоснящаяся кожа;
- разболтанность всех суставов.
Малые критерии:
- рецидивирующие дислокации;
- хронические артралгии;
- наличие родственников со схожими симптомами;
- недостаточность митрального клапана;
- дилятация корня аорты.
АД наследование
Сосудистый тип
Большие критерии:
- тонкая светлая кожа;
- разрывы сосудов;
- слабость мышц тазового дна;
- предрасположенность к образованию синяков;
- типичное лицо (утонченный заостренный нос, маленькие губы, кожа кажется натянутой, запавшие щеки, а из-за атрофии подкожной клетчатки глаза кажутся выкаченными).
Наличие двух или нескольких больших критериев убедительно свидетельствует об этом типе болезни.
Малые критерии:
- разболтанность малых суставов;
- травмы связочного и мышечного аппаратов;
- разрыв мочевого пузыря;
- конско-варусная стопа;
- варикозное расширение вен;
- артерио-венозные шунты и каротидно-кавернозное соустье;
- пневмо- или пневмогемоторакс;
- обнажение шеек и корней зубов;
- отягощенный семейный анамнез.
АД наследование. Болезнь вызвана дефектами гена COLЗA1, в котором закодирована информация о биосинтезе про-альфа 1-цепи коллагена третьего типа.
Суставной тип (артрохолазия)
Большие критерии:
- сильная разболтанность большинства суставов;
- повторяющиеся подвывихи;
- врожденный вывих бедра.
Малые критерии:
- чрезмерная растяжимость кожи, ранимость тканей;
- склонность к образованию синяков;
- мышечная гипотония;
- кифосколиоз;
- остеопороз.
АД наследование. Болезнь вызвана патологией генов COL1A1 и COL1A2, кодирующих альфа 1- и альфа2-цепи коллагена первого типа.
Кожный тип (дерматоспараксия)
Большие критерии:
- легкая ранимость кожи;
- обвисающая складчатая кожа.
Малые критерии:
- мягкая рыхлая кожа;
- предрасположенность к образованию синяков;
- дородовое излитие околоплодных вод;
- пупочная и паховая грыжи.
АР наследование. Больные — либо гомозиготы, либо смешанные гетерозиготы по мутации гена ADAMTS2, кодирующего проколлаген-N-эндопептидазу. Несмотря на лёгкую травмируемость кожи и склонность к образованию синяков, заживление ран происходит нормально.
Кифосколиотический тип
Большие критерии:
- разболтанность большинства суставов;
- тяжёлая мышечная гипотония у новорождённых;
- врождённый сколиоз, имеющий прогрессирующее развитие;
- ранимость склеры и разрыв глазного яблока.
Для верификации диагноза грудным детям необходимо присутствие 3 больших критериев.
Малые критерии:
- ранимость тканей;
- склонность к образованию синяков;
- разрывы артерий;
- марфаноподобная внешность;
- микрокорнеа;
- остеопороз.
АР наследование. В основе болезни лежит мутация гена лизилгидроксилазы (PLOD)-фермента, осуществляющего посттрансляционную модификацию проколлагена.
Многие больные к 20-30 годам теряют способность самостоятельно передвигаться.
Осложнения синдрома Элерса — Данлоса
- Самыми частыми осложнениями синдрома Элерса-Данло являются массивные кровотечения при разрывах крупных сосудов.
- При кифосколиотическом типе в достаточно раннем возрасте происходит утеря функции самостоятельного передвижения, связанная с патологией позвоночника. При этом же типе часто наступает потеря зрения из-за истончения оболочек глаза, приводящего к его разрыву.
Диагностика синдрома Элерса — Данлоса
Диагностика СЭД основывается на определении больших и малых критериев различных форм данного заболевания.
Объем исследований детерминируется присутствием главных клинических признаков заболевания. Существенное значение имеют сбор информации о семейной истории, проведение молекулярных исследований ДНК.
Для правильной верификации необходимо выполнять определенные правила:
- для постановки диагноза требуется присутствие минимум 1 большого критерия;
- при возможности лабораторного подтверждения анализа присутствие двух и более больших критериев гарантирует его подтверждение;
- малый критерий является симптомом, характеризующимся меньшей диагностической специфичностью. Присутствие малых критериев помогает определить тип синдрома;
- при невыявлении больших критериев малые не являются достаточными для верификации диагноза. Из-за того, что малые критерии встречаются у больных синдромом Элерса-Данло гораздо чаще, чем большие, наличие только малых критериев определяет возможность для верификации элерсоподобного фенотипа.
Дифференциальная диагностика проводится с другими видами дисплазии соединительной ткани. В случае невыполнения соответствующих критериев гипермобильность суставов должна расцениваться как самостоятельное состояние.
Для подтверждения диагноза дерматопараксазии проводят электрофорез коллагена первого типа, полученного из биоптата кожи или культуры фибробластов, в присутствии ингибиторов протеаз. Диагноз подтверждается при обнаружении нерасщепленных предшественников альфа 1- и альфа 2-цепей коллагена первого типа.
Лечение синдрома Элерса — Данлоса
При решении проблемы терапии больных СЭД большое внимание уделяется разработке вопросов немедикаментозного лечения — режим, диета, физиотерапия, психотерапия. Преимущественно немедикаментозные методы воздействия положены в основу существующих программ диспансерного учета больных.
Рекомендуется адекватная двигательная активность, лечебно-физкультурные мероприятия, массажи, физиолечение, ортопедическая реабилитация, адекватный выбор профессии. Высока эффективность гидротерапии, плавания.
Физиотерапию применяют при наличии соответствующих показаний. Достаточно широко используют магнитотерапию, индуктотерапию и применяют лечебный лазер. Физиотерапевтическое лечение детей со сколиозом, кифосколиозом должно включать КВЧ-терапию на болевые зоны (№10 каждый день или 1 раз в 2 дня), введение микроэлементов, спазмолитиков с помощью электрофоретических методов, амплипульсовые методы или диадинамотерапевтическую стимуляцию гипотоничных мышц; локальное лечение ультразвуком, курс вакуумного и ручного массажа, лечебную гимнастику и плавание, направленные на укрепление мышц.
Показано санаторное лечение, заключающееся в следующем:
- Режим щадяще-тренирующий. Необходимо исключить осевые перегрузки на позвоночник (поднятие тяжестей, прыжки, рывковые нагрузки);
- Массаж мышц спины релаксирующе-тонизирующий, № 10;
- Бальнеолечение (углекислосероводородные ванны по 10-12 мин, № 10);
- Пеллоидотерапия (грязевые аппликации) на мышцы вдоль позвоночника;
- ЛФК, специальный комплекс упражнений для укрепления мышц спины и плечевого пояса, часть которых проводится в положении лежа, чтобы исключить перегрузки позвоночника.
Применяется высокобелковая диета, наваристые бульоны, холодцы, заливные продукты. Курсы физиолечения, ЛФК. Лечение клинических проявлений, зависящее от степени поражения органов и систем. Медикаментозное лечение проводится с использованием аминокислотных (карнитин, нутраминос), витаминных (витамины D, C, E, B1, B2, B6), минеральных комплексов (магне В6, кальций-D3 никомед, магнерот), хондроитина перорально и местно, глюкозаминосульфата, оссеин-гидроксиапатитных комплексов (остеокеа, остеогенон, кальцимакс), трофических препаратов (АТФ, рибоксин, лецитин, кофермент Q10).
Указанные препараты принимаются сочетанными курсами 2-3 раза в 12 месяцев продолжительностью один-полтора мес.
Лекарственное симптоматическое лечение включает в себя купирование артралгии, миалгии, улучшение кровообращения, прием бета-блокеров, адаптогенных, успокоительных, вегетотропных препаратов.
Необходимо помнить, что СЭД является заболеванием с мультисистемным поражением, требуется разработка и практическое применение различных системных методов диагностики и лечения данной патологии.
Прогноз. Профилактика
Прогноз в большинстве случаев благоприятный, серьезнее он при первом (вследствие артропатий) и шестом (вследствие кровотечений и разрывов сосудов) типах. Детей следует ориентировать на выбор профессии, не связанной с физическими нагрузками, работой стоя.
Профилактика заключается в пренатальном выявлении заболевания у плода методом молекулярно-генетического анализа, прерывании беременности по медицинским показаниям. Постнатальная профилактика заболевания не разработана.
Профилактика осложнений включает в себя правильную и своевременную диагностику болезни и её лечение.
Список литературы
- Кеннет Л.Джонс Наследственные синдромы по Дэвиду Смиту Изд.«Практика», М., 2011. 1002 с.
- Козлова C.Наследственные синдромы и медико-генетическое консультирование. М.:Практика, 2007, 448 с.
- Barabas AP:Ehlers-Danlos syndrome. Associated with prematirity and premature of foetal membranes:possible increase in incidence. BMJ 2:682, 1966
- Бадалян Л.О, Скворцов И.А. Клиническая электронейромиография. Москва. «Медицина». – 1986. – 367с
- Синдром Элерса-Данлоса. В кн.: Наследственная патология человека. Под общ. Ред. Вельтищева Ю.Е., Бочкова Н.П. Москва. – 1992. — т.1. – с.100-109
- Barohn R.G. Distal myopathies and dystrophies // Smin. – Neurol. – 1993. – Sep. – 13 (3). – p. 247-55
- Барашнев Ю.И., Шехонин Б.В., Семячкина А.Н., Маккаев Х.М. Диагностика синдрома Элерса-Данло у детей. // Вопросы охраны материнства и детства. – 1988. – т.33. — № 11. – с. 59-64
- Делягин В.М., Нарычева И.А., Пильх А.Д. Синдром Элерса – Данлоса у детей // Педиатрия . – 1988. — № 12. – с. 8-15
- Ильина Н.А., Нарычева И.А., Аверьянов Ю.Н., Логунова Л.В. Клиническая и генетическая гетерогенность синдрома Элерса-Данлоса//Журн. невропатологии и психиатрии им. С.С.Корсакова. 1983. № 10. – с.1445- 1449




